We held the final AGM for the Marine Biosecurity Toolbox project, after five years of intensive research and collaborations.
Lessons from an international cross-laboratory experiment on delivering reproducible metabarcoding data
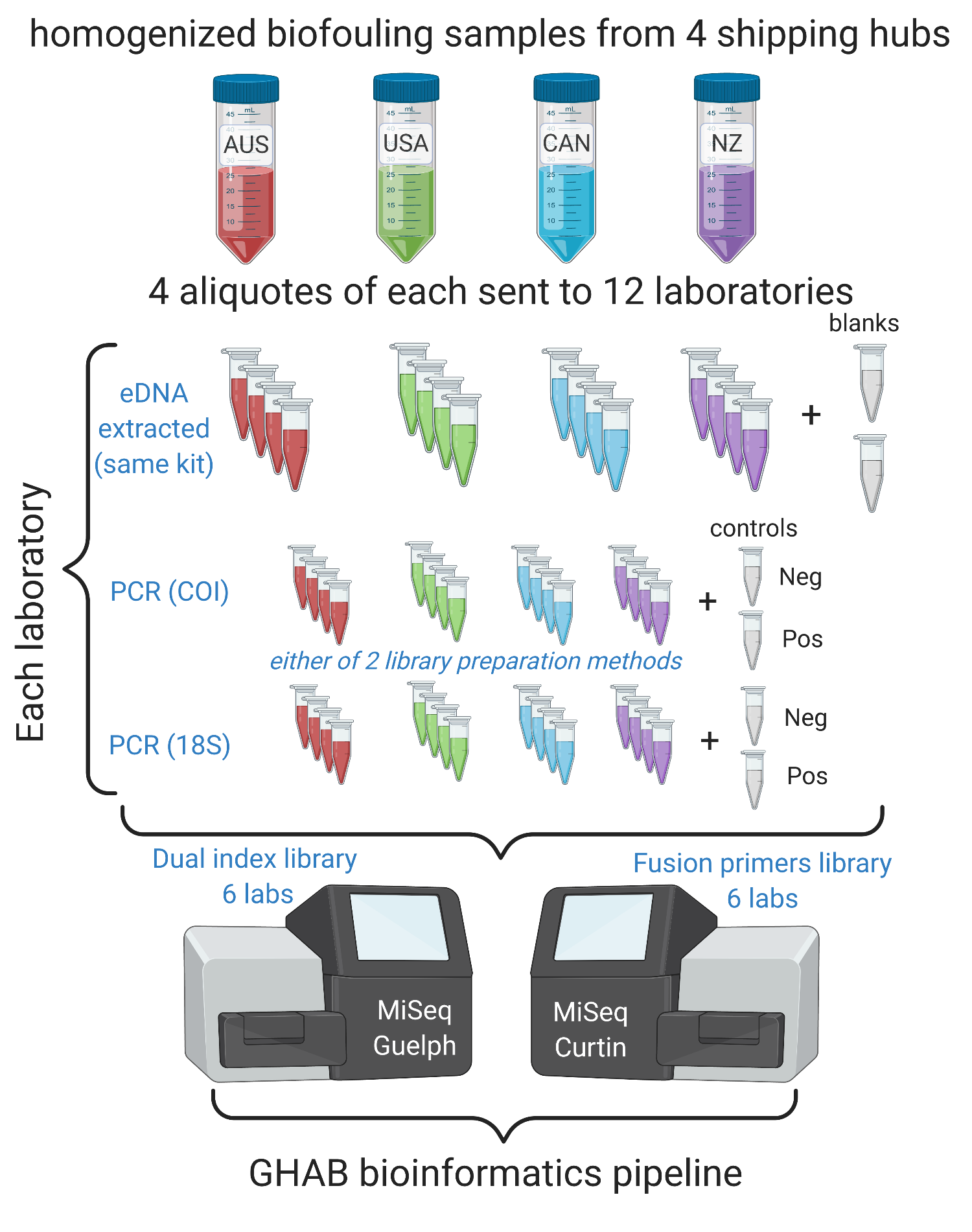
High-throughput sequencing (HTS) technologies are revolutionizing our ability to characterize biodiversity across ecosystems and offer unprecedented opportunities for biosecurity surveillance. Research institutions worldwide increasingly employ HTS methods for biodiversity assessments. However, variance in laboratory procedures, analytical workflows and bioinformatic pipelines impede the transferability and comparability of results across research groups. To assess the consistency of metabarcoding results derived from identical samples and primer sets using varying laboratory procedures, a unique cross-laboratory experiment was conducted as part of a Quadrilateral Scientific Collaboration. The results of this international effort are presented in the recent publication led by the DETECT team. The applied experimental design allowed us to identify discrepancies caused by laboratory-specific variation in technical steps and find the weakest links in the analytical pipelines
that resulted in the greatest divergence in metabarcoding data. This study presents some valuable empirical insights for standardizing future metabarcoding applications.
Related Posts



Comments (0)